MYCN
Gene Summary
| Gene: | MYCN; MYCN proto-oncogene, bHLH transcription factor |
| Aliases: | NMYC, ODED, MODED, N-myc, bHLHe37 |
| Location: | 2p24.3 |
| Summary: | This gene is a member of the MYC family and encodes a protein with a basic helix-loop-helix (bHLH) domain. This protein is located in the nucleus and must dimerize with another bHLH protein in order to bind DNA. Amplification of this gene is associated with a variety of tumors, most notably neuroblastomas. Multiple alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jun 2014] |
| Databases: | OMIM, HGNC, Ensembl, GeneCard, Gene |
| Protein: | N-myc proto-oncogene protein |
| Source: | NCBIAccessed: 01 September, 2019 |
Ontology: | What does this gene/protein do? Show (17) |
Contents
Gene Summary
Cancer Overview
Specific Cancers (9)
Useful Links
Latest Publications
Cancer Overview
MYCN is over-expressed in a number of different types of cancer, most notably neuroblastoma, but also inclusing rhabdomyosarcoma, medulloblastoma, astrocytoma, Wilms' tumour, and small cell lung cancer. In neuroblastoma MYCN amplification is an established indicator of poor-prognosis. MYCN belongs to a family of similar transcription factors (including C-MYC).
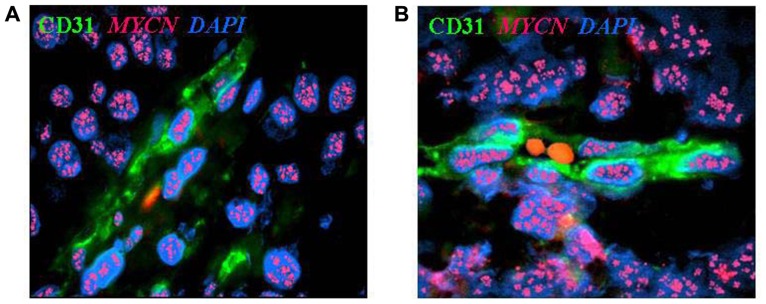
Neuroblastoma-derived endothelial micro-vessels. (A) Immunofluorescence and fluorescent in situ hybridization analysis of NB tumor section highlights CD31+ endothelial micro-vessel (green) carrying MYCN amplification (multiple red signals). (B) Two RBCs are in the open lumen of the NB-derived endothelial micro-vessel. Nuclei are stained with DAPI (blue), ×100.
Image from: from Pistoia et al, MYCN: from oncoprotein to tumor-associated antigen Front Oncol. 2012; 2: 174. License: CC BY 3.0.
Research Indicators
Graph generated 01 September 2019 using data from PubMed using criteria.
Literature Analysis
Mouse over the terms for more detail; many indicate links which you can click for dedicated pages about the topic.
Specific Cancers (9)
Data table showing topics related to specific cancers and associated disorders. Scope includes mutations and abnormal protein expression.
| Entity | Topic | PubMed | Papers |
| Neuroblastoma | MYCN amplification in NeuroblastomaPrognostic Amplification (duplicate copies) of the MYCN gene (also referred to as N-MYC) is established as an adverse prognostic factor in neuroblastoma. The amplicon (material co-amplified with MYCN) varies between patients, but can include the DDX1 gene. MYCN amplification is also correlates with 1p36 deletion and gain of chromosome 17q. MYCN amplified cell lines also overexpress ID2. |  View Publications View Publications | 1364 |
| Retinoblastoma | MYCN Amplification in Retinoblastoma There have been a number of reports of MYCN amplification in retinoblastoma cell lines and primary tumours. Doz et al. (1996) found only 1 of 45 primary retinoblastomas studies exhibited MYCN amplification while Kim et al. (1999) found 6 of 33 primary tumours were amplified. The latter study also suggested that MYCN amplified tumours have higher proliferation levels than non-amplified tumours. | View Publications | 113 |
| Rhabdomyosarcoma | MYCN Amplification in Rhabdomyosarcoma MYCN gene amplification occurs in a subset patients with rhabdomyosarcoma. It has been reported in 40-60% of patients with alveolar rhabdomyosarcoma, but infrequently occurs in patients with embyronal rhabdomyosarcoma. Some studies suggest that MYCN amplification in may be associated with a worse prognosis in rhabdomyosarcoma, however, findings have generally been based on small numbers. | View Publications | 61 |
| Lung Cancer | MYCN in Lung Cancer | View Publications | 51 |
| Medulloblastoma | MYCN Amplification in Medulloblastoma | View Publications | 28 |
| Osteosarcoma | MYCN Amplification in Osteosarcoma | View Publications | 13 |
| Neuroblastoma | ABCC1 (MRP1) Overexpression in Neuroblastoma Overexpression of MRP1 has been reported to have prognostic significance in neuroblastoma (Haber, 2006). There is also evidence that MRP1 is a target of MYCN (Blanc, 2003 & Manohar, 2004), frequently amplified in neuroblastoma, and involved in the development of multidrug resistance. | View Publications | 10 |
| Wilms Tumour | MYCN amplification in Wilms Tumor Williams et al (2010) reported MYCN amplification in 9% of cases in a SIOP study of over 100 Wilms tumor patients. This study also found that FBXW7 was mutated or deleted in approximately 4% of cases and the authors note that MYCN is a target of FBXW7-mediated ubiquitination and degradation - suggesting a common pathway is dysregulated by different mechanisms in various Wilms tumor subtypes. | View Publications | 10 |
| Breast Cancer | MYCN and Breast Cancer | View Publications | 4 |
Note: list is not exhaustive. Number of papers are based on searches of PubMed (click on topic title for arbitrary criteria used).
Useful Links

Atlas of Genetics and Cytogenetics in Oncology and Haematology
 MYCN
MYCN
OMIM, Johns Hopkin University
Referenced article focusing on the relationship between phenotype and genotype.
 MYCN
MYCN
International Cancer Genome Consortium.
Summary of gene and mutations by cancer type from ICGC
MYCN
Cancer Genome Anatomy Project, NCI
Gene Summary
 MYCN
MYCN
COSMIC, Sanger Institute
Somatic mutation information and related details
MYCN
GEO Profiles, NCBI
Search the gene expression profiles from curated DataSets in the Gene Expression Omnibus (GEO) repository.
Latest Publications: MYCN (cancer-related)
Targeting the Difficult-to-Drug CD71 and MYCN with Gambogic Acid and Vorinostat in a Class of Neuroblastomas.
Cell Physiol Biochem. 2019; 53(1):258-280 [PubMed] Related Publications
METHODS: Microarray analysis of cohorts of neuroblastoma patients indicated a subset of neuroblastomas overexpressing both CD71 and MYCN. The viability with proliferation changes were measured by MTT and colony formation assays in neuroblastoma cells. Transfection with CD71 or MYCN along with quantitative real-time polymerase chain reaction (qRT-PCR) and Western blotting were used to detect expression changes. For pathway analysis, gene ontology (GO) and Protein-protein interaction analyses were performed to evaluate the potential mechanisms of GA and vorinostat in treated cells.
RESULTS: For both GA and vorinostat, their pathways were explored for specificity and dependence on their targets for efficacy. For GA-treated cells, the viability/proliferation loss due to GA was dependent on the expression of CD71 and involved activation of caspase-3 and degradation of EGFR. It relied on the JNK-IRE1-mTORC1 pathway. The drug vorinostat also reduced cell viability/proliferation in the treated cells and this was dependent on the presence of MYCN as MYCN siRNA transfection led to a blunting of vorinostat efficacy and conversely, MYCN overexpression improved the vorinostat potency in those cells. Vorinostat inhibition of MYCN led to an increase of the pro-apoptotic miR183 levels and this, in turn, reduced the viability/proliferation of these cells. The combination treatment with GA and vorinostat synergistically reduced cell survival in the MYCN and CD71 overexpressing tumor cells. The same treatment had no effect or minimal effect on HEK293 and HEF cells used as models of non-cancer cells.
CONCLUSION: A combination therapy with GA and vorinostat may be suitable for MYCN and CD71 overexpressing neuroblastomas.
Current management of neuroblastoma and future direction.
Crit Rev Oncol Hematol. 2019; 138:38-43 [PubMed] Related Publications
Neuroblastoma
Downregulation of the histone methyltransferase SETD2 promotes imatinib resistance in chronic myeloid leukaemia cells.
Cell Prolif. 2019; 52(4):e12611 [PubMed] Related Publications
MATERIALS AND METHODS: The level of SETD2 in imatinib-sensitive and imatinib-resistant chronic myeloid leukaemia (CML) cells was examined by immunoblotting and quantitative real-time PCR. We analysed CD34
RESULTS: SETD2 was found to act as a tumour suppressor in CML. The novel oncogenic targets MYCN and ERG were shown to be the direct downstream targets of SETD2, where their overexpression induced by SETD2 knockdown caused imatinib insensitivity and leukaemic stem cell enrichment in CML cell lines. Treatment with JIB-04, an inhibitor that restores H3K36me3 levels through blockade of its demethylation, successfully improved the cell imatinib sensitivity and enhanced the chemotherapeutic effect.
CONCLUSIONS: Our study not only emphasizes the regulatory mechanism of SETD2 in CML, but also provides promising therapeutic strategies for overcoming the imatinib resistance in patients with CML.
Risk-modeling of dog osteosarcoma genome scans shows individuals with Mendelian-level polygenic risk are common.
BMC Genomics. 2019; 20(1):226 [PubMed] Free Access to Full Article Related Publications
RESULTS: First, meta-analysis revealed association near FGF9, which has strong biological and therapeutic relevance. Secondly, risk-modeling by multiple logistic regression shows 22 of the 34 associated loci contribute to risk and eight have large effect sizes. We validated the Greyhound stepwise model in our own, independent, case-control cohort. Lastly, we updated the gene annotation from approximately 50 genes to 175, and prioritized those using cross-species genomics data. Mostly positional evidence suggests 13 genes are likely to be associated with mapped risk (including MTMR9, EWSR1 retrogene, TANGO2 and FGF9). Previous annotation included seven of those 13 and prioritized four by pathway enrichment. Ten of our 13 priority genes are in loci that contribute to risk modeling and thus can be studied epidemiologically and translationally in pet dogs. Other new candidates include MYCN, SVIL and MIR100HG.
CONCLUSIONS: Polygenic osteosarcoma-risk commonly rises to Mendelian-levels in some dog breeds. This justifies caninized animal models and targeted clinical trials in pet dogs (e.g., using CDK4/6 and FGFR1/2 inhibitors).
Bone Cancers Osteosarcoma
The small molecule Bcl-2/Mcl-1 inhibitor TW-37 shows single-agent cytotoxicity in neuroblastoma cell lines.
BMC Cancer. 2019; 19(1):243 [PubMed] Free Access to Full Article Related Publications
METHODS: Cell viability, apoptosis, proliferation and changes in growth properties were determined in SKNAS, IMR-5, SY5Y and Kelly cells after treatment with TW-37. After transfection with Mcl-1 or Bcl-2 siRNA, apoptosis and proliferation were investigated in Kelly cells. Mice with Kelly cell line xenografts were treated with TW-37 and tumor growth, survival and apoptosis were determined.
RESULTS: Cell lines with N-Myc amplification were more sensitive to TW-37 treatment, IC50 values for IMR-5 and Kelly cells being 0.28 μM and 0.22 μM, compared to SY5Y cells and SKNAS cells (IC50 0.96 μM and 0.83 μM). Treatment with TW-37 resulted in increased apoptosis and reduced proliferation rates, especially in IMR5 and Kelly cells. Bcl-2 as well as Mcl-1 knockdown induced apoptosis in Kelly cells. TW-37 led to a decrease in tumor growth and a favorable survival (p = 0.0379) in a Kelly neuroblastoma xenografts mouse model.
CONCLUSION: TW-37 has strong single-agent cytotoxicity in vitro and in vivo. Therefore, combined inhibition of Bcl-2/Mcl-1 by TW-37 in N-Myc amplified neuroblastoma may represent an interesting therapeutic strategy.
Targeting excessive MYCN expression using MLN8237 and JQ1 impairs the growth of hepatoblastoma cells.
Int J Oncol. 2019; 54(5):1853-1863 [PubMed] Related Publications
ACC1 is overexpressed in liver cancers and contributes to the proliferation of human hepatoma Hep G2 cells and the rat liver cell line BRL 3A.
Mol Med Rep. 2019; 19(5):3431-3440 [PubMed] Free Access to Full Article Related Publications
Liver Cancer
Metallothionein 3 Is a Hypoxia-Upregulated Oncogene Enhancing Cell Invasion and Tumorigenesis in Human Bladder Carcinoma Cells.
Int J Mol Sci. 2019; 20(4) [PubMed] Free Access to Full Article Related Publications
Neuroblastoma of the Iris in Children.
J Pediatr Ophthalmol Strabismus. 2019; 56:e12-e16 [PubMed] Related Publications
Neuroblastoma
Inhibition of N-myc expression sensitizes human neuroblastoma IMR-32 cells expressing caspase-8 to TRAIL.
Cell Prolif. 2019; 52(3):e12577 [PubMed] Related Publications
MATERIALS AND METHODS: We established N-myc-downregulated IMR-32 cells using shRNA lentiviral particles targeting N-myc and examined the effect the N-myc inhibition on TRAIL susceptibility in human neuroblastoma IMR-32 cells expressing caspase-8.
RESULTS: Cisplatin treatment in IMR-32 cells increased the expression of death receptor 5 (DR5; TRAIL-R2), but not other receptors, via downregulation of NF-κB activity. However, the cisplatin-mediated increase in DR5 failed to induce cell death following TRAIL treatment. Furthermore, interferon (IFN)-γ pretreatment increased caspase-8 expression in IMR-32 cells, but cisplatin failed to trigger TRAIL cytotoxicity. We downregulated N-myc expression in IMR-32 cells using N-myc-targeting shRNA. These cells showed decreased growth rate and Bcl-2 expression accompanied by a mild collapse in the mitochondrial membrane potential as compared with those treated with scrambled shRNA. TRAIL treatment in N-myc-negative cells expressing caspase-8 following IFN-γ treatment significantly triggered apoptotic cell death. Concurrent treatment with cisplatin enhanced TRAIL-mediated cytotoxicity, which was abrogated by an additional pretreatment with DR5:Fc chimera protein.
CONCLUSIONS: N-myc and caspase-8 expressions are involved in TRAIL susceptibility in IMR-32 cells, and the combination of treatment with cisplatin and TRAIL may serve as a promising strategy for the development of therapeutics against neuroblastoma that is controlled by N-myc and caspase-8 expression.
Basic Gene Expression Characteristics of Glioma Stem Cells and Human Glioblastoma.
Anticancer Res. 2019; 39(2):597-607 [PubMed] Related Publications
MATERIALS AND METHODS: Total RNA was extracted from GSCs, U251 and GBM and microarray analysis was performed; the data were then applied to the bioinformatics pipeline consisting of a principal component analysis (PCA) with factor loadings, an intracellular pathway analysis, and an immunopathway analysis.
RESULTS: The PCA clearly distinguished the three groups. The factor loadings of the PCA suggested that v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog (MYCN), dipeptidyl-peptidase 4 (DPP4), and macrophage migration-inhibitory factor (MIF) contribute to the stemness of GSCs. The intracellular pathway and immunopathway analyses provided relevant information about the functions of representative genes in GSCs.
CONCLUSION: The newly-developed cellular bioinformatics pipeline was a useful method to clarify the similarities and differences among samples.
Droplet digital PCR as an alternative to FISH for MYCN amplification detection in human neuroblastoma FFPE samples.
BMC Cancer. 2019; 19(1):106 [PubMed] Free Access to Full Article Related Publications
METHODS: In the present study, we examined the concordance of droplet digital PCR (ddPCR, in combination with immunohistochemistry, IHC) with FISH for MYCN detection in a panel of formalin-fixed paraffin-embedded (FFPE) human neuroblastoma samples.
RESULTS: In 112 neuroblastoma cases, ddPCR analysis demonstrated a 96-100% concordance with FISH. Consistently, IHC grading revealed 92-100% concordance with FISH. Comparing ddPCR with IHC, we observed a concordance of 95-98%.
CONCLUSIONS: The results demonstrate that MYCN amplification status in NB cases can be assessed with ddPCR, and suggest that ddPCR could be a technically less challenging method of detecting MYCN status in FFPE specimens. More importantly, these findings illustrate the concordance between FISH and ddPCR in the detection of MYCN status. Together, the results suggest that rapid, less technically demanding, and inexpensive ddPCR in conjunction with IHC could serve as an alternate approach to detect MYCN status in NB cases, with near-identical sensitivity to that of FISH.
FISH Neuroblastoma
Crocin inhibits proliferation and induces apoptosis through suppressing MYCN expression in retinoblastoma.
J Biochem Mol Toxicol. 2019; 33(5):e22292 [PubMed] Related Publications
Apoptosis Retinoblastoma
Stage 4 s neuroblastoma: features, management and outcome of 268 cases from the Italian Neuroblastoma Registry.
Ital J Pediatr. 2019; 45(1):8 [PubMed] Free Access to Full Article Related Publications
METHODS: Of 3355 subjects aged 0-18 years with previously untreated neuroblastoma diagnosed between 1979 and 2013, a total of 280 infants (8.3%) had stage 4 s characteristics, 268 of whom were eligible for analyses. Three treatment eras were identified on the basis of based diagnostic and chemotherapy adopted. Group 1 patients received upfront chemotherapy; Group 2 and 3 patients underwent observation in the absence of life-threatening symptoms (LTS), except for Group 3 patients with amplified MYCN gene, who received more aggressive therapy.
RESULTS: The three groups were comparable, with few exceptions. Ten-year overall survival significantly increased from 76.9 to 89.7% and was worse for male gender, age 0-29 days and presence of selected LTS on diagnosis, elevated LDH, and abnormal biologic features. Infants who underwent primary resection ± chemotherapy did significantly better. On multivariate analysis, treatment eras and the association of hepatomegaly to dyspnea were independently associated with worse outcome.
CONCLUSIONS: Our data confirm that stage 4 s neuroblastoma is curable in nearly 90% of cases. Hepatomegaly associated to dyspnea was the most important independent risk factor. The cure rate could be further increased through timely identification of patients at risk who might benefit from surgical techniques, such as intra-arterial chemoembolization and/or liver transplantation, which must be carried out in institutions with specific expertise.
Neuroblastoma
Predicting clinical outcomes in neuroblastoma with genomic data integration.
Biol Direct. 2018; 13(1):20 [PubMed] Related Publications
RESULTS: Our supervised model trained on the SEQC cohort can accurately predict overall survival and event-free survival profiles of patients in two independent cohorts. We also performed extensive experiments to assess the prediction accuracy of high risk patients and patients without MYCN amplification. Our results from this part suggest that clinical endpoints can be predicted accurately across multiple cohorts. To explore the data in an unsupervised manner, we used an integrative clustering strategy named multi-view kernel k-means (MVKKM) that can effectively integrate multiple high-dimensional datasets with varying weights. We observed that integrating different gene expression datasets results in a better patient stratification compared to using these datasets individually. Also, our identified subgroups provide a better Cox regression model fit compared to the existing risk group definitions.
CONCLUSION: Altogether, our results indicate that integration of multiple genomic characterizations enables the discovery of subtypes that improve over existing definitions of risk groups. Effective prediction of survival times will have a direct impact on choosing the right therapies for patients.
REVIEWERS: This article was reviewed by Susmita Datta, Wenzhong Xiao and Ziv Shkedy.
Neuroblastoma
Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma.
Oncogene. 2019; 38(15):2800-2813 [PubMed] Free Access to Full Article Related Publications
ALK positively regulates MYCN activity through repression of HBP1 expression.
Oncogene. 2019; 38(15):2690-2705 [PubMed] Related Publications
Nobiletin Enhances Chemosensitivity to Adriamycin through Modulation of the Akt/GSK3β/β⁻Catenin/MYCN/MRP1 Signaling Pathway in A549 Human Non-Small-Cell Lung Cancer Cells.
Nutrients. 2018; 10(12) [PubMed] Free Access to Full Article Related Publications
MYCN is amplified during S phase, and c‑myb is involved in controlling MYCN expression and amplification in MYCN‑amplified neuroblastoma cell lines.
Mol Med Rep. 2019; 19(1):345-361 [PubMed] Free Access to Full Article Related Publications
E2F1 Transcription Factor Neuroblastoma
PPP3CB contributes to poor prognosis through activating nuclear factor of activated T-cells signaling in neuroblastoma.
Mol Carcinog. 2019; 58(3):426-435 [PubMed] Related Publications
Apoptosis Neuroblastoma
TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets.
Nat Commun. 2018; 9(1):4866 [PubMed] Free Access to Full Article Related Publications
JARID2 Functions as a Tumor Suppressor in Myeloid Neoplasms by Repressing Self-Renewal in Hematopoietic Progenitor Cells.
Cancer Cell. 2018; 34(5):741-756.e8 [PubMed] Article available free on PMC after 12/11/2019 Related Publications
Acute Myeloid Leukemia (AML)
Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors.
Nat Commun. 2018; 9(1):4760 [PubMed] Article available free on PMC after 12/11/2019 Related Publications
MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis.
Mol Cell. 2018; 72(5):836-848.e7 [PubMed] Related Publications
Meta-mining of copy number profiles of high-risk neuroblastoma tumors.
Sci Data. 2018; 5:180240 [PubMed] Article available free on PMC after 12/11/2019 Related Publications
Neuroblastoma
Two siblings with familial neuroblastoma with distinct clinical phenotypes harboring an ALK germline mutation.
Genes Chromosomes Cancer. 2018; 57(12):665-669 [PubMed] Related Publications
miR‑501‑3p sensitizes glioma cells to cisplatin by targeting MYCN.
Mol Med Rep. 2018; 18(5):4747-4752 [PubMed] Related Publications
Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors.
Proc Natl Acad Sci U S A. 2018; 115(40):E9391-E9400 [PubMed] Article available free on PMC after 12/11/2019 Related Publications
Retinoblastoma RB1
Innate immune sensor laboratory of genetics and physiology 2 suppresses tumor cell growth and functions as a prognostic marker in neuroblastoma.
Cancer Sci. 2018; 109(11):3494-3502 [PubMed] Article available free on PMC after 12/11/2019 Related Publications
Neuroblastoma
Molecular and epidemiologic characterization of Wilms tumor from Baghdad, Iraq.
World J Pediatr. 2018; 14(6):585-593 [PubMed] Article available free on PMC after 01/12/2019 Related Publications
METHODS: Next-generation sequencing of ten target genes associated with WT development and treatment resistance (WT1, CTNNB1, WTX, IGF2, CITED1, SIX2, p53, N-MYC, CRABP2, and TOP2A) was completed. Immunohistochemistry was performed for 6 marker proteins of WT (WT1, CTNNB1, NCAM, CITED1, SIX2, and p53). Patient outcomes were compiled.
RESULTS: Mutations were detected in previously described WT "hot spots" (e.g., WT1 and CTNNB1) as well as novel loci that may be unique to the Iraqi population. Immunohistochemistry showed expression domains most typical of blastemal-predominant WT. Remarkably, despite the challenges facing families and care providers, only one child, with combined WT1 and CTNNB1 mutations, was confirmed dead from disease. Median clinical follow-up was 40.5 months (range 6-78 months).
CONCLUSIONS: These data suggest that WT biology within a population of Iraqi children manifests features both similar to and unique from disease variants in other regions of the world. These observations will help to risk stratify WT patients living in this difficult environment to more or less intensive therapies and to focus treatment on cell-specific targets.
Disclaimer: This site is for educational purposes only; it can not be used in diagnosis or treatment.
Cite this page: Cotterill SJ. MYCN (n-myc), Cancer Genetics Web: http://www.cancer-genetics.org/MYCN.htm Accessed:

This page in Cancer Genetics Web by Simon Cotterill is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Note: content of abstracts copyright of respective publishers - seek permission where appropriate.
[Home] Page last revised: 01 September, 2019 Cancer Genetics Web, Established 1999